Как правильно установить строительные леса
Категории блога
Популярные записи
15 ноября 2021 г.
Возведение зданий из кладочных материалов, отделка фасадов, монтаж различных коммуникаций на высоте — всё это и многое другое требует наличия строительных лесов. Данное оборудование позволяет рабочим свободно перемещаться вдоль внешних или внутренних стен на любом из имеющихся уровней и тем самым обеспечивает им удобство при выполнении профессиональных задач. Изготавливаются леса на соответствующих предприятиях. На крупных объектах монтируются с привлечением спецтехники. В домашних условиях допускается сборка своими силами и даже изготовление из подручных материалов, что значительно сокращает денежные затраты. Но в таких случаях велики риски обрушения конструкций и травмирования людей. Поэтому, чтобы помочь желающим сэкономить и в то же время избежать неприятностей, предлагаем несколько минут уделить вопросу «Как правильно установить строительные леса?».
Из каких элементов собираются строительные леса
Строительные леса, будь то заводские или самодельные, собираются из определённого набора комплектующих. Основными среди них считаются:
Основными среди них считаются:
- вертикальные стойки, являющиеся базой для крепления остальных элементов и обеспечивающие конструкции необходимую высоту;
- упоры, служащие основаниями для стоек и препятствующие заглублению последних в грунт;
- диагональные и горизонтальные распорки, соединяющие стойки и обеспечивающие леса необходимой стабильностью;
- перемычки, выполняющие функции оснований для настилов и увеличивающие прочность всей конструкции;
- лестницы, размещающиеся на боковых сторонах лесов и дающие возможность строителям подниматься на требуемый ярус или опускаться на землю;
- сварные крюки, шурупы-кольца и прочие детали, играющие роль соединительных элементов и позволяющие фиксировать все комплектующие в необходимом положении;
- настилы, опирающиеся на перемычки и представляющие собой платформы для размещения мастеров, инструментов, материалов;
- ограждения в виде бортиков или поручней, обрамляющие настилы и препятствующие выпадению рабочих и инвентаря.


Также могут использоваться кронштейны, которые позволяют выполнять крепление лесов к сооружению, домкратные системы, которые упрощают выравнивание составляющих по горизонтали, винтовые опоры, которые делают конструкцию регулируемой, сетчатая ткань, которая защищает мастеров и обрабатываемые поверхности от агрессивных влияний ветра, осадков, солнечных лучей. Их количество, размеры и список в целом варьируются в зависимости от этажности строящегося или подвергающегося ремонту здания, конфигурации стен, особенностей грунта или искусственного покрытия в зоне расположения отмосток и от ряда других факторов. При этом отправной точкой в процессе подбора комплектующих является определение актуального для конкретной ситуации типа конструкции.
Какими бывают конструкции строительных лесов
Принадлежность обсуждаемого в данной статье оборудования к той или иной группе по типу конструкции влияет не только на его комплектацию, но и на решение вопроса о том, как собираются строительные леса.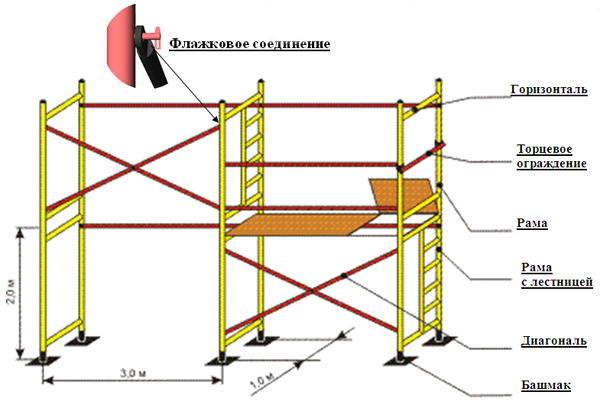 Поэтому самое время ознакомиться с разновидностями последних, которых не так уж и мало.
Поэтому самое время ознакомиться с разновидностями последних, которых не так уж и мало.
- Рамные конструкции из жёстких металлических секций прямоугольной формы, соединяющихся между собой специальными распорками. Используются при выполнении строительных, отделочных, реставрационных и других работ внутри и снаружи зданий.
- Штыревые
- Хомутовые каркасные конструкции с креплениями в виде хомутов. Обладают возможностью регулировать шаг стоек и ярусную высоту, хорошо комбинируются с другими типами лесов, а потому подходят для строительства или отделки зданий сложной конфигурации, как, например, водонапорных башен или соборов с куполами.
- Клиновые леса с клиновыми соединениями замков и фланцев. Позволяют возводить, ремонтировать или реконструировать сооружения сложной или нестандартной формы — крытые павильоны, навесы, сцены, трибуны и прочее. Подходят для применения как снаружи, так и внутри зданий.
- Модульные системы с компактной конструкцией. Используются при сооружении или отделке объектов цилиндрической и сферической формы — башен, котлов, мостов. Могут достигать высоты 60 м.
- Башенные леса, собираемые из тех же элементов, что и модульные. Отличаются деталями, дающими возможность выполнять перемещения параллельно фасадной площади.
- Вышки-туры, имеющие конструкцию башенного типа. Обладают высокой мобильностью. Используются при выполнении внутренних или наружных работ на высоте до 20 м.
- Подвесные системы, опирающиеся на соответствующие консоли. Незаменимы на участках, расположенных над ветхими козырьками или пристройками.
- Леса-термосы, оснащённые плёнкой для сохранения температуры подогреваемого изнутри воздуха. Применяются в случаях, когда строительные или отделочные работы проводятся в холодное время года.
 Позволяют возводить, ремонтировать или реконструировать сооружения сложной или нестандартной формы — крытые павильоны, навесы, сцены, трибуны и прочее. Подходят для применения как снаружи, так и внутри зданий.
Позволяют возводить, ремонтировать или реконструировать сооружения сложной или нестандартной формы — крытые павильоны, навесы, сцены, трибуны и прочее. Подходят для применения как снаружи, так и внутри зданий. Применяются в случаях, когда строительные или отделочные работы проводятся в холодное время года.
Применяются в случаях, когда строительные или отделочные работы проводятся в холодное время года.Наибольшей популярностью среди перечисленных модификаций пользуются рамные леса, так как в них наилучшим образом сочетаются простота монтажа, небольшой вес, высокая прочность, универсальность применения и доступная цена. Плюс и в том, что данное оборудование не вызывает особых трудностей даже в сборке из элементов, изготовленных своими руками. А вот как устанавливать такие строительные леса, рассмотрим далее.
Как правильно собирать строительные леса рамного типа
Сборка и установка рамных, как и любых других, лесов начинается с зачистки, выравнивания и трамбовки прилегающей к зданию или сооружению территории шириной не менее 3 м. План проведения дальнейших работ выглядит следующим образом:
- Размещение на земле вдоль стен деревянных подкладок толщиной 4 см. Если основание ровное и достаточно твёрдое, например, асфальтированное или бетонированное, то такие подкладки не требуются.
- Выставление подпятников (башмаков) в точках, предназначенных для размещения опорных элементов.
- Закрепление в башмаках двух смежных секций.
- Соединение этих рам горизонтальными и диагональными связями посредством флажковых замков.
- Поочерёдное выставление и закрепление остальных секций первого ряда по принципу, описанному в пунктах 3 и 4.
- Наращивание получившейся конструкции до высоты первого яруса путём применения метода «труба в трубу». При этом рамы второго и всех последующих рядов соединяются между собой так же, как и в первом.
- Установка ригелей.
- Укладывание и фиксация настилов. При образовании щитов из продольно расположенных досок просветы между последними оставляются шириной не более 0,5 см. Стыки и нахлёсты размещаются на опорных элементах. Борта делаются высотой не менее 15 см.
- Монтаж второго яруса по схеме, заключённой в шагах 6–8.
- Фиксация второго яруса на стене здания с помощью анкерных кронштейнов.
- Сборка остальных ярусов в последовательности, описанной в пунктах 6–10.


Вполне возможно, что для решения ваших задач больше подходит конструкция, отличная от рамной. В таком случае о том, как собрать и закрепить строительные леса, узнаете из соответствующей инструкции. Она в обязательном порядке включается в комплект как нового, так и сдающегося в аренду оборудования.
Монтаж строительных лесов в Москве
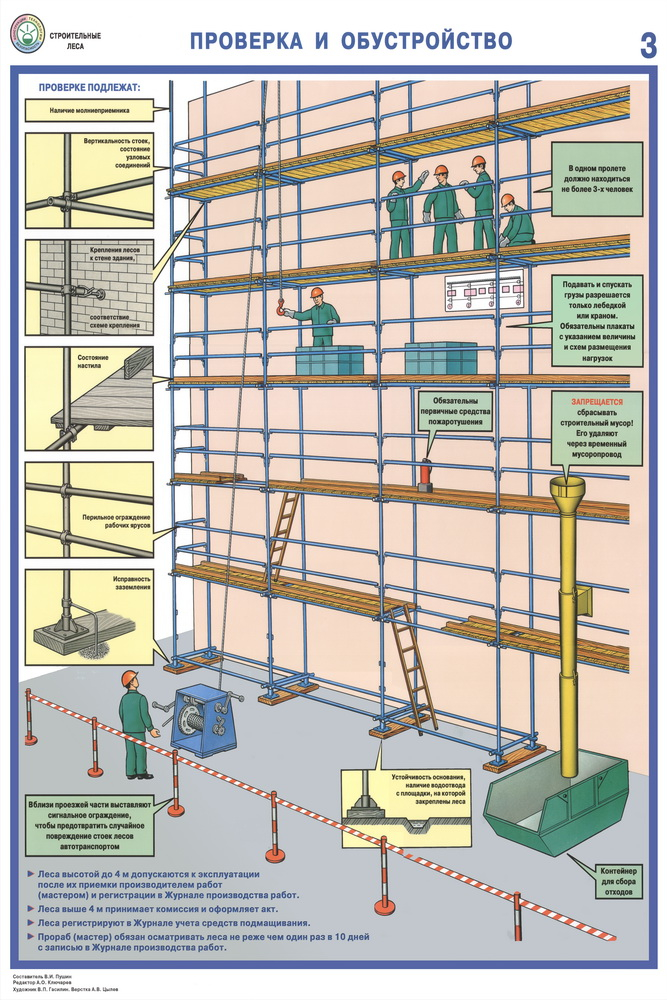
Строительные леса – популярное оборудование для строительства, отделки или ремонта на высоте от 2-х метров. Металлоконструкции доставляются на объект в разобранном виде, они должны быть смонтированы в соответствии с правилами и требованиям стандартов безопасности.
ООО «Алеся Д» предоставляет услуги профессионального монтажа и демонтажа строительных лесов на объектах в Москве и Московской области.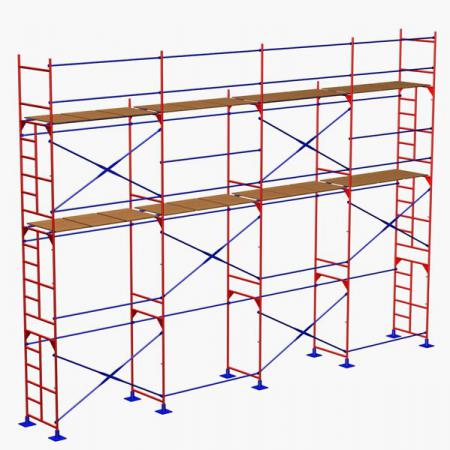 Наши сотрудники строго соблюдают все технологические требования, проводят контроль качества сборки лесов и устойчивости высотной металлоконструкции.
Наши сотрудники строго соблюдают все технологические требования, проводят контроль качества сборки лесов и устойчивости высотной металлоконструкции.
Стоимость услуг
Цена монтажа строительных лесов зависит от типа и конфигурации металлоконструкций, срочности и других параметров. Стоимость услуги просчитывается индивидуально в каждом конкретном случае.
Последовательность монтажа
Перед сборкой лесов необходимо подготовить площадку. Она должна быть ровной, плотной и без перепадов высоты.
Строительные леса устанавливаются в несколько этапов:
- монтаж опорных пят;
- установка рам первого яруса с горизонтальными и диагональными связями;
- монтаж второго и последующего ярусов с креплением к фасаду здания;
- сооружение настилов на выбранных ярусах лесов.
Специалисты нашей компании обладают высоким уровнем квалификации и имеют допуски для сборки высотных конструкций. Монтажные работы проводятся под руководством ответственного сотрудника, рабочие обеспечены всеми необходимыми приспособлениями безопасности.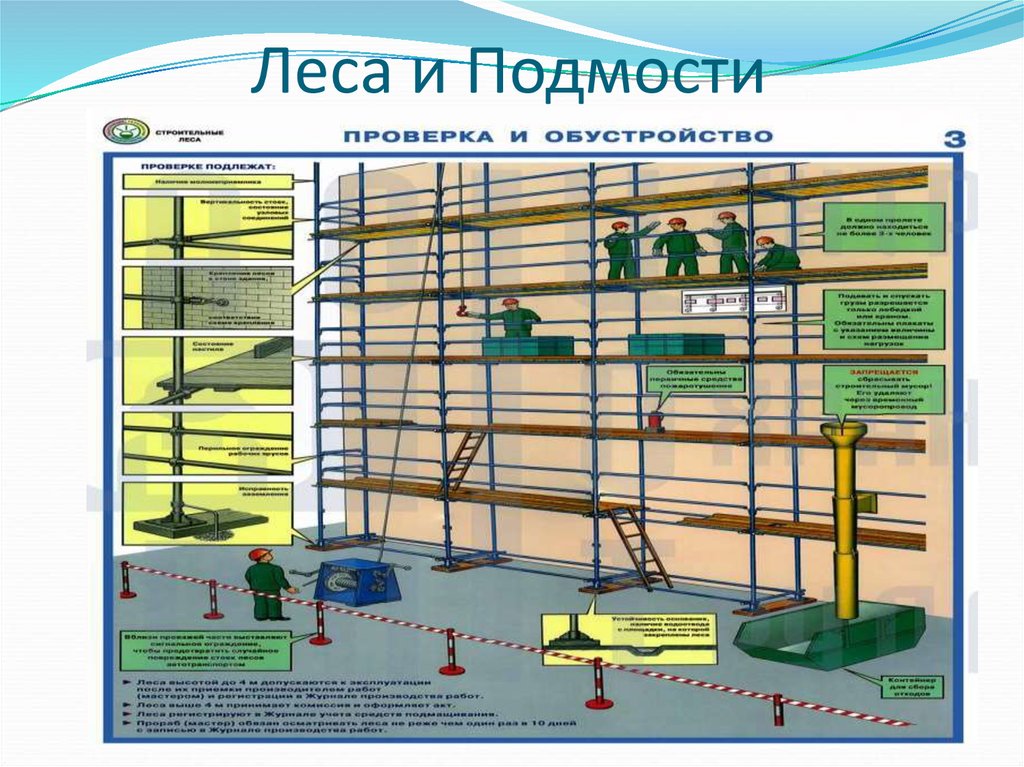
Преимущества компании
ООО «Алеся Д» гарантирует качественный монтаж строительных лесов любого типа и конфигурации:
- оперативный выезд на объект;
- доступные цены;
- контроль качества сборки.
Мы можем смонтировать ваши леса или предоставить оборудование в аренду. Установка проводится в строгом соответствии со строительными нормами и правилами, схемами и инструкцией производителя. Мы установили доступные расценки на услуги и предлагаем гибкую систему скидок.
Как оставить заявку
Чтобы заказать услугу монтажа высотных лесов, отправьте заявку в онлайн чате или свяжитесь с нами по телефону +7 (495) 741-32-26. Наш специалист оперативно выедет на объект для оценки объема, сложности и стоимости работ.
В оговоренное время к вам прибудет бригада монтажников, которая выполнит сборку и установку металлоконструкций. По завершении составляется акт выполненных работ, который подписывается исполнителем и заказчиком.
Отзывы о нас
Евгений Петрович Хворостов, Одинцово
Мы с моим сыном строили дачу. Первый этаж быстро отстроили, а вот со вторым заминка вышла. Пытались с помощью подручных приспособлений работать, да на высоте это не очень удобно
Анатолий Сидорчук, предпринимательЯ владелец развивающейся строительной фирмы, всем необходимым оборудованием обзавелся не сразу. Обратился в компанию «Алеся Д».
Читать все отзывы
Более подробную информацию о продаже строительных лесов Вы можете получить, оставив вопрос в форме обратной связи,
выслав нам свой запрос по электронной почте [email protected]
или по телефонам (495) 741-32-26, (495) 111-57-07
Мы будем рады ответить на все интересующие Вас вопросы.
ntJoin: быстрое и легкое построение шаблонов на основе сборки с использованием графов минимизации | Биоинформатика
Статья журнала
Лорен Кумб,
Лорен Кумб
Ищите другие работы этого автора на:
Оксфордский академический
пабмед
Google Scholar
Владимир Николич,
Ищите другие работы этого автора на:
Оксфордский академический
пабмед
Google Scholar
Джастин Чу,
Джастин Чу
Ищите другие работы этого автора на:
Оксфордский академический
пабмед
Google Scholar
Инанк Бироль,
Инанк Бирол
Ищите другие работы этого автора на:
Оксфордский академический
пабмед
Google Scholar
Рене Л. Уоррен
Уоррен
Рене Л. Уоррен
Ищите другие работы этого автора на:
Оксфордский академический
пабмед
Google Scholar
Биоинформатика , Том 36, Выпуск 12, 15 июня 2020 г., Страницы 3885–3887, https://doi.org/10.1093/bioinformatics/btaa253
Опубликовано:
3 20042 209 004 20003 История статьи
Получено:
13 января 2020 г.
Получена редакция:
23 марта 2020 г.
Принято:
14 апреля 2020 г. PDF
- Содержание статьи
- Рисунки и таблицы
- видео
- Аудио
- Дополнительные данные
Цитировать
Cite
Lauren Coombe, Vladimir Nikolić, Justin Chu, Inanc Birol, René L Warren, ntJoin: Быстрые и легкие строительные леса, управляемые сборкой, с использованием минимизирующих графов, Bioinformatics , Volume 36, Issue 12, 15 June 2020, Pages 3885 –3887, https://doi.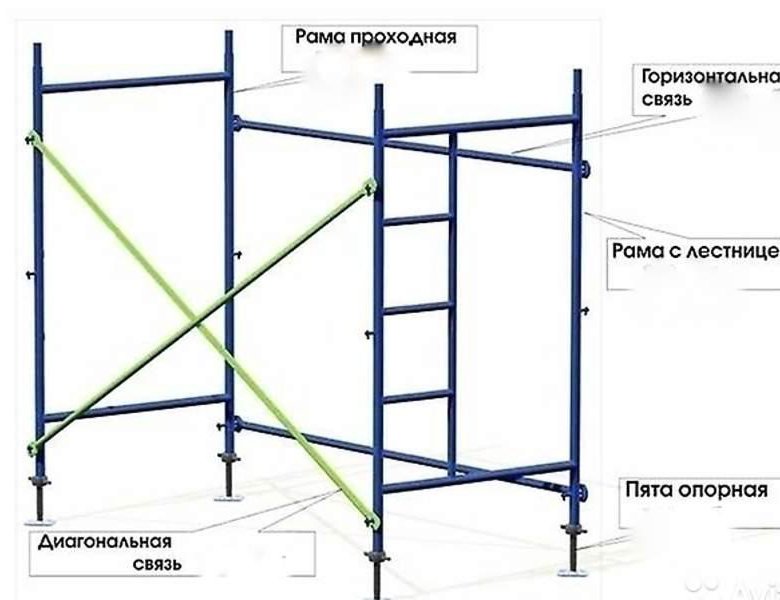 org/10.1093/bioinformatics/btaa253
org/10.1093/bioinformatics/btaa253
Выберите формат Выберите format.ris (Mendeley, Papers, Zotero).enw (EndNote).bibtex (BibTex).txt (Medlars, RefWorks)
Закрыть
Разрешения
- Электронная почта
- Твиттер
- Подробнее
Фильтр поиска панели навигации БиоинформатикаЭтот выпускЖурналы по биоинформатикеБиоинформатика и вычислительная биологияКнигиЖурналыOxford Academic Термин поиска мобильного микросайта
Закрыть
Фильтр поиска панели навигации БиоинформатикаЭтот выпускЖурналы по биоинформатикеБиоинформатика и вычислительная биологияКнигиЖурналыOxford Academic Термин поиска на микросайте
Advanced Search
Abstract
Summary
Возможность генерировать высококачественные последовательности генома является краеугольным камнем современных биологических исследований. Даже с недавними достижениями в технологиях секвенирования многие сборки генома все еще не достигают эталонного уровня. Здесь мы представляем ntJoin, инструмент, который использует структурную синтению между черновой сборкой и ссылочной последовательностью(ями) для сопряжения и исправления первой по отношению к последней. Вместо выравнивания ntJoin использует упрощенный подход к сопоставлению, основанный на структуре данных графа, созданной из упорядоченных эскизов минимизатора. Этот инструмент можно использовать в различных приложениях, в том числе для улучшения черновой сборки с помощью генома эталонного уровня, сборки для короткого чтения с черновой сборкой для длинного чтения и черновой сборки для сборки из близкородственного вида. При построении сборки короткого считывания человека с использованием эталонного генома человека или сборки длинного считывания ntJoin улучшает длину NGA50 в 23 и 13 раз соответственно при длине менее 13 м и использовании менее 11 ГБ ОЗУ. По сравнению с существующими скаффолдерами, основанными на ссылках, ntJoin создает очень непрерывные сборки быстрее и использует меньше памяти.
Даже с недавними достижениями в технологиях секвенирования многие сборки генома все еще не достигают эталонного уровня. Здесь мы представляем ntJoin, инструмент, который использует структурную синтению между черновой сборкой и ссылочной последовательностью(ями) для сопряжения и исправления первой по отношению к последней. Вместо выравнивания ntJoin использует упрощенный подход к сопоставлению, основанный на структуре данных графа, созданной из упорядоченных эскизов минимизатора. Этот инструмент можно использовать в различных приложениях, в том числе для улучшения черновой сборки с помощью генома эталонного уровня, сборки для короткого чтения с черновой сборкой для длинного чтения и черновой сборки для сборки из близкородственного вида. При построении сборки короткого считывания человека с использованием эталонного генома человека или сборки длинного считывания ntJoin улучшает длину NGA50 в 23 и 13 раз соответственно при длине менее 13 м и использовании менее 11 ГБ ОЗУ. По сравнению с существующими скаффолдерами, основанными на ссылках, ntJoin создает очень непрерывные сборки быстрее и использует меньше памяти.
Доступность и реализация
ntJoin написан на C++ и Python и находится в свободном доступе на https://github.com/bcgsc/ntjoin.
Дополнительная информация
Дополнительные данные доступны по адресу Биоинформатика онлайн.
1 Введение
Создание сильно непрерывных сборок позволяет проводить важные последующие исследования, такие как исследования генетических ассоциаций и анализ цис--регуляторных элементов (Rice and Green, 2019). Однако, несмотря на то, что продвижение данных секвенирования отдельных молекул, таких как связанные чтения и длинные чтения, показало большие перспективы в улучшении качества сборки генома de novo (Shafin et al. , 2019; Weisenfeld et al. , 2017), большинство черновых сборок все еще не достигают полноты хромосомного масштаба.
Для некоторых черновиков геномов могут быть доступны более смежные сборки для разных особей того же вида или даже близкородственных видов. В этом случае синхронизация последовательности между сборками может быть использована для построения лесов, управляемых сборкой. Например, в то время как сборки с длительным чтением могут генерировать непрерывные черновики геномов, высокая частота ошибок чтения отрицательно влияет на базовое качество, затрудняя аннотацию генов (Watson and Warr, 2019).). Полировка с использованием коротких считываний часто используется для повышения точности сборки в паре оснований (Rice and Green, 2019; Warren et al. , 2019; Watson and Warr, 2019). Альтернативный подход к этому шагу полировки состоит в том, чтобы собрать короткие риды отдельно и построить сборку коротких ридов, используя сборку длинных ридов, создавая сборку наравне с непрерывностью и структурой сборки длинных ридов и точностью пары оснований сборка короткого чтения.
В этом случае синхронизация последовательности между сборками может быть использована для построения лесов, управляемых сборкой. Например, в то время как сборки с длительным чтением могут генерировать непрерывные черновики геномов, высокая частота ошибок чтения отрицательно влияет на базовое качество, затрудняя аннотацию генов (Watson and Warr, 2019).). Полировка с использованием коротких считываний часто используется для повышения точности сборки в паре оснований (Rice and Green, 2019; Warren et al. , 2019; Watson and Warr, 2019). Альтернативный подход к этому шагу полировки состоит в том, чтобы собрать короткие риды отдельно и построить сборку коротких ридов, используя сборку длинных ридов, создавая сборку наравне с непрерывностью и структурой сборки длинных ридов и точностью пары оснований сборка короткого чтения.
Существующие опорные леса, такие как Ragout (Колмогоров и др. , 2018 г.) и Ragoo (Alonge et al. , 2019 г.) полагаются на выравнивание черновой сборки с эталонной сборкой; Ragout использует Progressive Cactus (Armstrong et al. , 2019) для больших геномов, в то время как Ragoo использует для этой задачи minimap2 (Li, 2018). Использование набросков минимизатора в таких инструментах, как minimap2, очень эффективно для компактного представления последовательностей генома. Вместо того, чтобы сохранять каждое слово размера k (k-мер) из входных последовательностей, сохраняется только выбранный набор k-меров или хэш-значений («минимизаторы»), что значительно снижает вычислительные затраты на хранение данных последовательности и манипулирование ими (Робертс и др. , 2004).
, 2019) для больших геномов, в то время как Ragoo использует для этой задачи minimap2 (Li, 2018). Использование набросков минимизатора в таких инструментах, как minimap2, очень эффективно для компактного представления последовательностей генома. Вместо того, чтобы сохранять каждое слово размера k (k-мер) из входных последовательностей, сохраняется только выбранный набор k-меров или хэш-значений («минимизаторы»), что значительно снижает вычислительные затраты на хранение данных последовательности и манипулирование ими (Робертс и др. , 2004).
Здесь мы представляем ntJoin, скаффолдер, управляемый сборкой, который использует упрощенную стратегию сопоставления без выравнивания вместо выравнивания для быстрого сопряжения целевой сборки с использованием одной или нескольких ссылок.
2 Материалы и методы
Имея входную целевую и эталонную последовательности в формате fasta, ntJoin сначала создает упорядоченный эскиз минимизатора для каждого из предоставленных наборов последовательностей, как описано ранее (Roberts et al.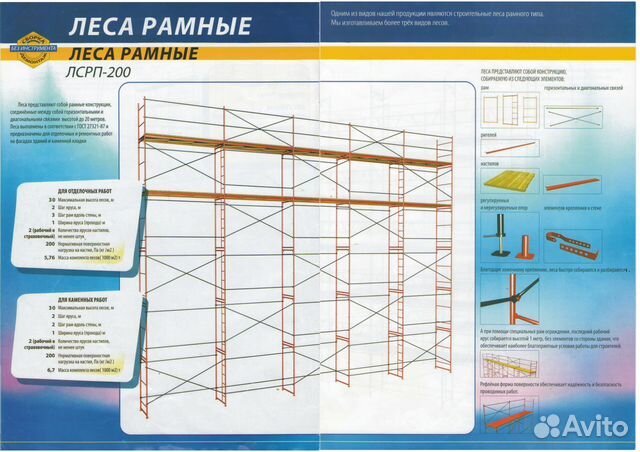 , 2004) (дополнительные рисунки S1 и S2). Затем ntJoin использует упорядоченные эскизы минимизатора из каждого входа для построения единого неориентированного графа, который облегчает легкое сопоставление между ними. В этом графе каждый узел является минимайзером, а ребра создаются между минимизаторами, соседними хотя бы в одном из упорядоченных эскизов. Вес ребра — это мера, которая используется для того, чтобы сделать больший акцент на соединениях в определенных входных сборках. Веса ребер представляют собой сумму заданных пользователем весов каждого входа, который поддерживает это ребро.
, 2004) (дополнительные рисунки S1 и S2). Затем ntJoin использует упорядоченные эскизы минимизатора из каждого входа для построения единого неориентированного графа, который облегчает легкое сопоставление между ними. В этом графе каждый узел является минимайзером, а ребра создаются между минимизаторами, соседними хотя бы в одном из упорядоченных эскизов. Вес ребра — это мера, которая используется для того, чтобы сделать больший акцент на соединениях в определенных входных сборках. Веса ребер представляют собой сумму заданных пользователем весов каждого входа, который поддерживает это ребро.
Затем граф подвергается серии шагов фильтрации. Во-первых, применяется глобальный порог веса ребра. Затем идентифицируются узлы ветвления (узлы со степенью > 2), и инцидентные ребра фильтруются с возрастающим пороговым значением веса ребра до тех пор, пока степень этого узла не упадет до <3. Как правило, вес эталона выше, чем вес целевой сборки, что приводит к тому, что эти ребра имеют приоритет, и в результате целевая сборка подгоняется к эталонной структуре. Фильтрация инцидентных ребер узлов ветвления приводит к тому, что граф представляет собой набор компонент связности, каждая из которых представляет собой линейный путь узлов-минимизаторов. Последовательности минимизаторов в линейных путях затем переводятся в упорядоченные и ориентированные непрерывные пути, которые описывают окончательные выходные каркасы (дополнительные рисунки S1 – S3). Этот основанный на графе метод позволяет алгоритму выполнять исправление неправильной сборки в дополнение к формированию входных контигов на основе входной эталонной сборки, поскольку контиги могут быть нарушены при предполагаемых ошибках сборки в режиме по умолчанию. Если пользователь не хочет, чтобы входные контиги обрезались при подгонке эталонной последовательности, можно указать параметр «no_cut=True».
Фильтрация инцидентных ребер узлов ветвления приводит к тому, что граф представляет собой набор компонент связности, каждая из которых представляет собой линейный путь узлов-минимизаторов. Последовательности минимизаторов в линейных путях затем переводятся в упорядоченные и ориентированные непрерывные пути, которые описывают окончательные выходные каркасы (дополнительные рисунки S1 – S3). Этот основанный на графе метод позволяет алгоритму выполнять исправление неправильной сборки в дополнение к формированию входных контигов на основе входной эталонной сборки, поскольку контиги могут быть нарушены при предполагаемых ошибках сборки в режиме по умолчанию. Если пользователь не хочет, чтобы входные контиги обрезались при подгонке эталонной последовательности, можно указать параметр «no_cut=True».
Окончательная целевая сборка в формате fasta является основным результатом работы ntJoin. Кроме того, сведения о том, как была создана целевая сборка, включая ориентацию, порядок и размеры зазоров, описываются в выходном файле «пути» и, необязательно, в файле agp (опция «agp = True»). Наконец, граф минимизатора выводится в «точечном» формате, который дает все узлы и ребра в графе, а также веса ребер, указывающие уровень поддержки сборки.
Наконец, граф минимизатора выводится в «точечном» формате, который дает все узлы и ребра в графе, а также веса ребер, указывающие уровень поддержки сборки.
Каждая сборка оценивалась на непрерывность и правильность с помощью QUAST (v5.0.2; –scaffold-gap-max-size 100 000 –large) (Михеенко и др. , 2018). Эта настройка параметра зазора каркаса приводит к тому, что несоответствия размера зазора более 100 КБ классифицируются как «обширные неправильные сборки».
Подробные методы доступны в Интернете.
3 Результаты и обсуждение
Сначала мы протестировали ntJoin с использованием различных сборок образцов Caenorhabditis elegans и Homo sapiens (дополнительные таблицы S1 и S2). По сравнению с Ragout и Ragoo, ntJoin обычно создает сборки с более высокой длиной NGA50 (длина, которая захватывает не менее 50% генома, используя длины выравнивания с эталоном вместо длин контигов) и сопоставимые или меньшие неправильные сборки (рис.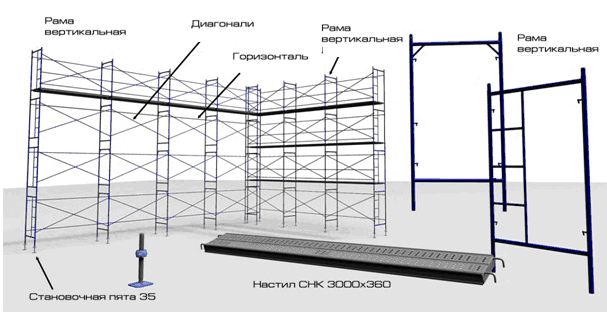 1; Дополнительные рисунки S4–S10; дополнительные таблицы S3–S11). Примечательно, что ntJoin улучшает сборки с начальной смежностью в диапазоне килобаз, чтобы достичь масштаба в мегабазах (NGA50 увеличивается с 26,9kb до 2,3 Мбит/с и от 19,8 kb до 50,3 Мбит/с для сборок с коротким чтением C.elegans и H.sapiens , соответственно, дополнительные рисунки S4 и S6, при этом количество неправильных сборок сократилось более чем на треть (33,5 и 61,5% , соответственно). Это подчеркивает потенциал ntJoin в улучшении фрагментированных черновых сборок. По сравнению с Ragout, ntJoin достиг значений NGA50 в 1,1-2 раза выше для протестированных сборок ABySS с коротким считыванием, хотя Ragout действительно создал основу для сборки Shasta с длинным считыванием до 1,2-кратного увеличения NGA50 (рис. 1; дополнительные рисунки S4 и S6) (Джекман и др. , 2017; Шафин и др. , 2019). Однако выравнивание Progressive Cactus, необходимое для Ragout, было очень затратным с точки зрения вычислений, выполнялось более четырех дней для всех запусков вручную и использовало более 115 ГБ ОЗУ по сравнению с запусками ntJoin для людей, которые завершились менее чем за 13 минут и использовали <11 ГБ памяти.
1; Дополнительные рисунки S4–S10; дополнительные таблицы S3–S11). Примечательно, что ntJoin улучшает сборки с начальной смежностью в диапазоне килобаз, чтобы достичь масштаба в мегабазах (NGA50 увеличивается с 26,9kb до 2,3 Мбит/с и от 19,8 kb до 50,3 Мбит/с для сборок с коротким чтением C.elegans и H.sapiens , соответственно, дополнительные рисунки S4 и S6, при этом количество неправильных сборок сократилось более чем на треть (33,5 и 61,5% , соответственно). Это подчеркивает потенциал ntJoin в улучшении фрагментированных черновых сборок. По сравнению с Ragout, ntJoin достиг значений NGA50 в 1,1-2 раза выше для протестированных сборок ABySS с коротким считыванием, хотя Ragout действительно создал основу для сборки Shasta с длинным считыванием до 1,2-кратного увеличения NGA50 (рис. 1; дополнительные рисунки S4 и S6) (Джекман и др. , 2017; Шафин и др. , 2019). Однако выравнивание Progressive Cactus, необходимое для Ragout, было очень затратным с точки зрения вычислений, выполнялось более четырех дней для всех запусков вручную и использовало более 115 ГБ ОЗУ по сравнению с запусками ntJoin для людей, которые завершились менее чем за 13 минут и использовали <11 ГБ памяти. БАРАН. ntJoin также был быстрее Ragoo во всех тестах: от 1,4 раза быстрее для сборки Shasta до 35,8 раза быстрее для более фрагментированной сборки H.sapiens ABySS (рис. 1; дополнительные рисунки S4 и S6). ntПрисоединиться к местам 86,3–99,3% входной сборки в лесах, пропорция, которая очень похожа как на Ragout, так и на Ragoo. Последовательности не могут быть помещены в каркасы, если они слишком короткие по сравнению с установленным пользователем размером окна или если выбранная ссылка слишком расходится. В то время как Ragoo использует постоянный размер промежутка между соединяемыми контигами (по умолчанию 100 bp), ntJoin и Ragout оценивают размеры промежутка на основе эталона, о чем свидетельствует общий размер промежутка в каркасных сборках Ragoo, который значительно меньше, чем у ntJoin и Ragout (дополнительная информация). Таблицы S4–S15).
БАРАН. ntJoin также был быстрее Ragoo во всех тестах: от 1,4 раза быстрее для сборки Shasta до 35,8 раза быстрее для более фрагментированной сборки H.sapiens ABySS (рис. 1; дополнительные рисунки S4 и S6). ntПрисоединиться к местам 86,3–99,3% входной сборки в лесах, пропорция, которая очень похожа как на Ragout, так и на Ragoo. Последовательности не могут быть помещены в каркасы, если они слишком короткие по сравнению с установленным пользователем размером окна или если выбранная ссылка слишком расходится. В то время как Ragoo использует постоянный размер промежутка между соединяемыми контигами (по умолчанию 100 bp), ntJoin и Ragout оценивают размеры промежутка на основе эталона, о чем свидетельствует общий размер промежутка в каркасных сборках Ragoo, который значительно меньше, чем у ntJoin и Ragout (дополнительная информация). Таблицы S4–S15).
Рис. 1.
Открыть в новой вкладке Скачать слайд различные сборки H.sapiens (NA12878) в линейном (а) и логарифмическом масштабе (б).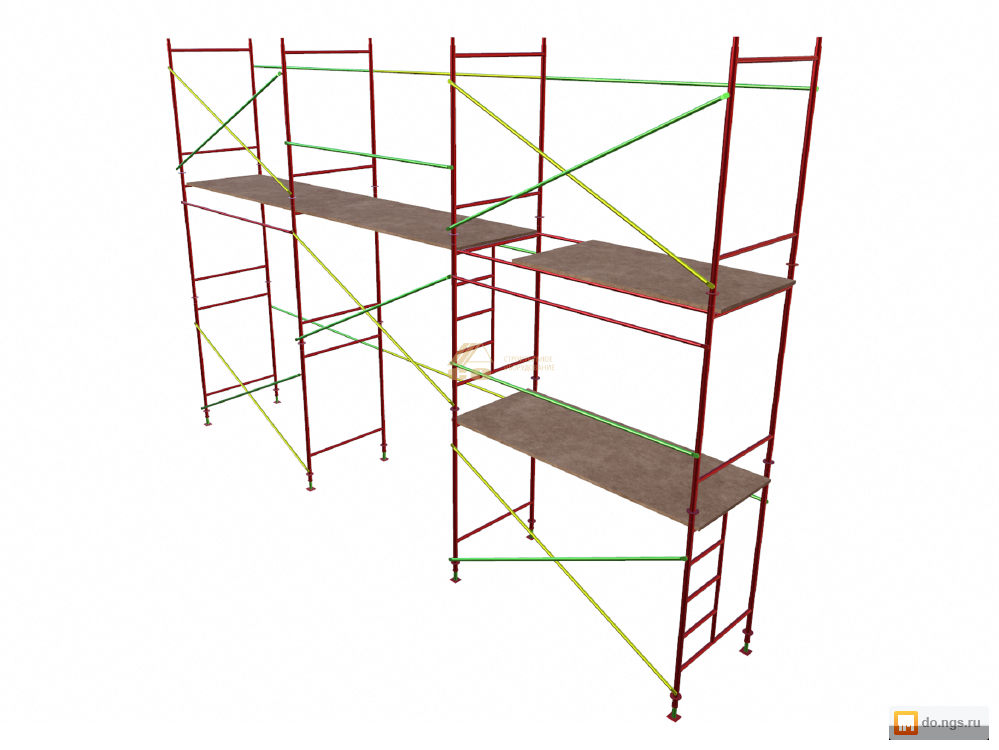 Эталонные геномы представляют собой эталонный геном человека («Ref») и отполированную с помощью ntEdit сборку Shasta («Shasta»). Улучшаемые целевые сборки — это сборка NA12878 ABySS, созданная с использованием данных MPET («ABySS»), и сборка Shasta, отполированная с помощью ntEdit («Shasta»). «Базовая» статистика показана для соответствующих целевых сборок до возведения лесов на каждой панели (а)
Эталонные геномы представляют собой эталонный геном человека («Ref») и отполированную с помощью ntEdit сборку Shasta («Shasta»). Улучшаемые целевые сборки — это сборка NA12878 ABySS, созданная с использованием данных MPET («ABySS»), и сборка Shasta, отполированная с помощью ntEdit («Shasta»). «Базовая» статистика показана для соответствующих целевых сборок до возведения лесов на каждой панели (а)
ntJoin также может улучшить черновики сборок, если для одного и того же вида доступны смежные сборки. Это типичный вариант использования в проекте, использующем несколько платформ секвенирования для гибридной сборки. На рисунке 1 сборка ABySS с коротким считыванием была построена с использованием сборки Shasta с длинным считыванием (Shafin et al. , 2019). Сохраняя соединения, уникальные для длинных и коротких последовательностей чтения, ntJoin достигает NGA50 выше, чем базовая сборка Shasta. ntJoin предоставляет альтернативный конвейер сборки, в котором структура сборки с длительным чтением сообщает о размещении последовательностей сборок с коротким чтением, исключая необходимость полировки сборки с длительным чтением с помощью коротких операций чтения.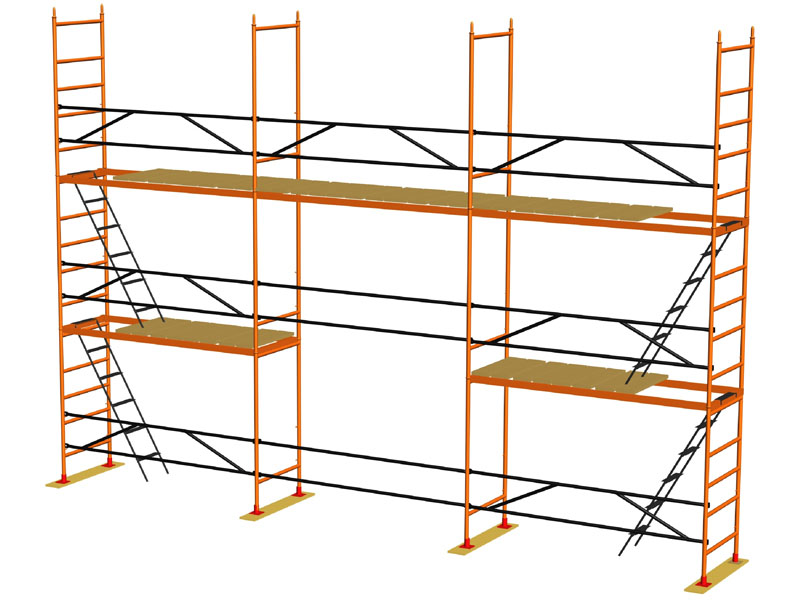 Этот подход может создавать сборки с высокой непрерывностью и базовой точностью, что особенно важно для аннотации нижестоящего генома (дополнительные таблицы S12–S15). Согласно этим данным, ни Ragout, ни Ragoo не дают сборок с одинаково высокой длиной NGA50, и обе требуют больше времени и памяти по сравнению с ntJoin.
Этот подход может создавать сборки с высокой непрерывностью и базовой точностью, что особенно важно для аннотации нижестоящего генома (дополнительные таблицы S12–S15). Согласно этим данным, ни Ragout, ни Ragoo не дают сборок с одинаково высокой длиной NGA50, и обе требуют больше времени и памяти по сравнению с ntJoin.
Подход ntJoin также распространяется на сборки лесов разных видов, как было продемонстрировано созданием лесов сборок морских и гавиальных крокодилов с использованием генома американского аллигатора в качестве эталона (дополнительные таблицы S16 и S17). В наших тестах длина NG50 крокодиловых сборок увеличилась до 14,44 и 12,92 Мбн для морских и гавиальных крокодилов (исходный уровень NG50 = 0,14 и 0,07 Мбп) с соответствующим увеличением полноты генов BUSCO (Simão et al. , 2015). 2,6 и 90,5% соответственно. Это показывает, что ntJoin все еще может использовать синтению между этими целевыми и эталонными сборками, несмотря на то, что виды разошлись около 80 миллионов лет назад (Delsuc et al. , 2018).
, 2018).
Любому инструменту сборки, основанному на эталонах, присуще некоторое смещение эталона, и пользователь должен учитывать это при разработке своего эксперимента, в том числе при выборе эталонной сборки. Здесь мы демонстрируем полезность ntJoin для подгонки входной целевой сборки к структуре эталона, что исправляет неправильные сборки, но также потенциально ломает некоторые большие структурные варианты. Точно так же инструменты компаратора были запущены в режимах, которые также могут обрезать входные контиги. Хотя этот режим не нарушит все структурные изменения (дополнительная таблица S18), чтобы избежать разрыва/обрезания входных контигов, можно указать параметр ntJoin no_cut = True, который предотвращает стирание любых существующих структурных изменений в целевой сборке (дополнительный рис. С11).
В заключение следует отметить, что ntJoin быстро и с небольшим объемом памяти выполняет минимизацию построения каркасов на основе графов, сохраняя при этом смежность на уровне хромосом. Как продемонстрировано, это гибкий, не требующий выравнивания инструмент построения каркаса, который можно использовать в ряде различных приложений, включая гибридную сборку и исследования популяционной геномики.
Как продемонстрировано, это гибкий, не требующий выравнивания инструмент построения каркаса, который можно использовать в ряде различных приложений, включая гибридную сборку и исследования популяционной геномики.
Финансирование
Эта работа была поддержана Genome BC и Genome Canada [243FOR, 281ANV]; и Национальных институтов здравоохранения [2R01HG007182-04A1]. Ответственность за содержание этой статьи несут исключительно авторы, и она не обязательно отражает официальную точку зрения Национальных институтов здравоохранения или других финансирующих организаций.
Конфликт интересов : не объявлено.
Ссылки
Аонге
М.
и др. (
2019
)
RaGOO: быстрое и точное построение черновиков геномов с использованием справочных материалов
.
Геном Биол
.,
20
,
17
.
Armstrong
J.
и др. (
2019
) Прогрессивное выравнивание с Cactus: мультигеномный выравниватель для эпохи тысяч геномов. биоРксив ,
730531
.
Delsuc
F.
и др. (
2018
)
Филогеномная основа и шкала времени для сравнительных исследований оболочников
.
БМС Биол
.,
16
,
39
.
Джекман
С.Д.
и др. (
2017
)
ABySS 2.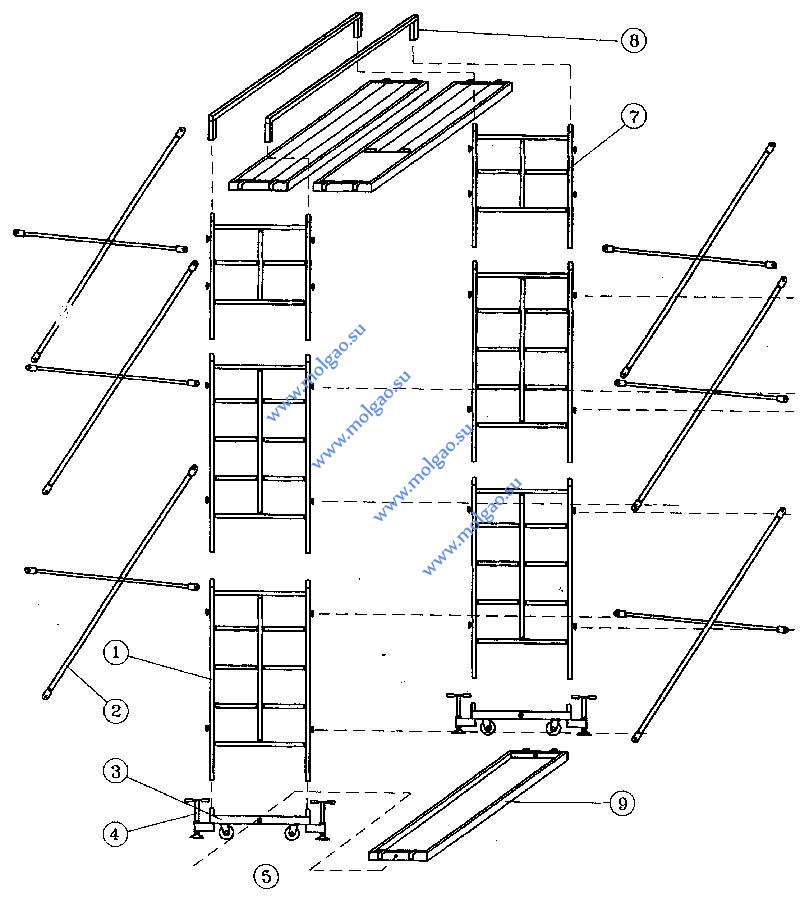 0: ресурсоэффективная сборка больших геномов с использованием фильтра Блума
0: ресурсоэффективная сборка больших геномов с использованием фильтра Блума
.
Геном Res
.,
27
,
768
–
777
.
Колмогоров
М.
и др. (
2018
)
Хромосомная сборка больших и сложных геномов с использованием нескольких ссылок
.
Геном Res
.,
28
,
1720
–
1732
.
Ли
Х.
(
2018
)
Minimap2: попарное выравнивание нуклеотидных последовательностей
.
Биоинформатика.
Михеенко
А.
и др. (
2018
)
Универсальная оценка сборки генома с помощью QUAST-LG
.
Биоинформатика
,
34
,
i142
–
i150
.
Рис
E.S.
,
Зеленый
Р.Е.
(
2019
)
Новые подходы к сборке генома и каркасу
.
год. Преподобный Аним. Biosci
.,
7
,
17
–
40
.
Робертс
М.
и др. (
2004
)
Снижение требований к хранению для сравнения биологических последовательностей
.
Биоинформатика.
Шафин
К.
и др. (
2019
) Эффективная сборка de novo одиннадцати геномов человека с использованием секвенирования PromethION и нового набора инструментов для нанопор. биоРксив ,
715722
.
Simão
F.A.
и др. (
2015
)
BUSCO: оценка сборки генома и полноты аннотации с помощью однокопийных ортологов
.
Биоинформатика.
Уоррен
Р.Л.
и др. (
(
2019
)
ntEdit: полировка масштабируемой последовательности генома
.
Биоинформатика.
Watson
М.
,
Уорр
А.
(
2019
)
Ошибки в сборках с длительным считыванием могут критически повлиять на предсказание белка
.
Нац. Биотехнолог
.,
37
,
124
–
126
.
Вайзенфельд
Н.И.
и др. (
2017
)
Прямое определение последовательностей диплоидного генома
.
Геном Res
.,
27
,
757
–
767
.
© Автор(ы), 2020. Опубликовано Oxford University Press.
Это статья в открытом доступе, распространяемая в соответствии с лицензией Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), которая разрешает неограниченное повторное использование, распространение и воспроизведение на любом носителе при условии, что оригинальная работа правильно цитируется.
Раздел выдачи:
ГЕНОМНЫЙ АНАЛИЗ
Помощник редактора: Альфонсо Валенсия
Альфонсо Валенсия
Ассоциированный редактор
Ищите другие работы этого автора на:
Оксфордский академический
пабмед
Google Scholar
Скачать все слайды
Дополнительные данные
Дополнительные данные
btaa253_Supplementary_Data – pdf файл
Реклама
Цитаты
Альтметрика
Дополнительная информация о метриках
Оповещения по электронной почте
Оповещение об активности статьи
Предварительные уведомления о статьях
Оповещение о новой проблеме
Получайте эксклюзивные предложения и обновления от Oxford Academic
Ссылки на статьи по телефону
Последний
Самые читаемые
Самые цитируемые
Систематический анализ данных альтернативного сплайсинга во времени с использованием Spycone
KMCP: точное метагеномное профилирование как прокариотических, так и вирусных популяций путем псевдокартирования
КРЕВЕТКИ: Компактные пангеномные функции для полногеномной популяционной геномики
Крупномасштабное прогнозирование белков, связанных с побочными реакциями на лекарства, с встраиванием в сеть
Несбалансированная классификация для субклеточной локализации белка с мультиметочной передискретизацией
Академический специалист по легочной медицине сна в живописной Центральной Пенсильвании
Херши, Пенсильвания
АССОЦИАЦИЯ/ПОЛНЫЙ ПРОФЕССОР – Кафедра расширенной эхокардиографической визуализации
Торонто, Онтарио
кандидат BC/BE
Бэй Шор, Нью-Йорк
Академический патолог-хирург/патолог молочной железы
, Вермонт
Просмотреть все вакансии
Реклама
Геномы против genNNN: разница между контигами и скаффолдами в сборках генома
3 февраля 2021 | Секвенирование 101
Этот пост в блоге был обновлен. Первоначально он был опубликован в сентябре 2016 года.
Первоначально он был опубликован в сентябре 2016 года.
В ходе недавних взаимодействий с научным сообществом мы стали свидетелями растущего числа вопросов, связанных с созданием каркасов геномных сборок. Мы подумали, что может быть полезно рассмотреть концепции, лежащие в основе контигов и каркасов, а также обстоятельства, при которых может потребоваться создание каркаса высококачественной сборки генома PacBio.
Контиги против каркасов
Контиги — это непрерывные участки последовательности, содержащие только основания A, C, G или T без пробелов. Секвенирование SMRT обладает всеми необходимыми характеристиками производительности — длинным чтением, отсутствием систематической ошибки последовательности и контекста и высокой точностью — для создания непрерывных сборок генома с контигами размером в мегануклеотиды. Сверхдлинные контиги предоставляют полную и непрерывную информацию о последовательностях полных генов, а в последнее время даже позволяют разделить разные хромосомы для диплоидных и полиплоидных организмов.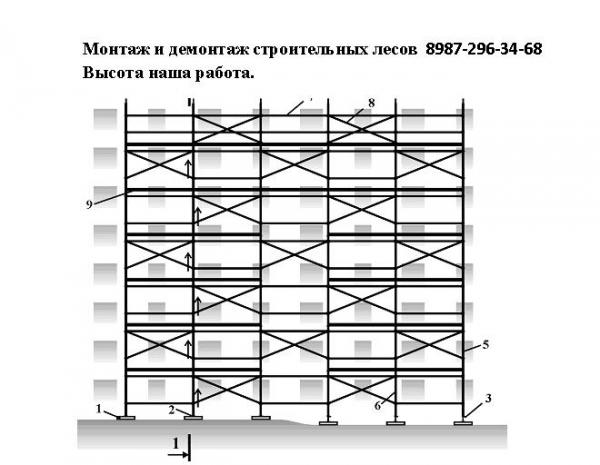
Беспрецедентное качество высокоточных длинных считываний PacBio, известных как считывания HiFi, было описано как «наиболее эффективная отдельная технология для сборки de novo » в исследовании, посвященном секвенированию клеточной линии человека CHM13, в результате которого был получен сборочный контиг. N50 размером 29,5 Мб и показателем качества Phred Q45. Чтения HiFi также позволили создать эталонное качество de novo сборок многих видов растений и животных, специфических для популяций человеческих сборок и первую полностью полную последовательность аутосом человека — хромосому 8, включая центромеры. Даже большие и сложные геномы растений, такие как калифорнийское красное дерево, гексаплоид размером 27 ГБ, могут быть легко собраны с высокой степенью непрерывности с помощью считывания HiFi.
Узнайте, как чтение HiFi помогает ученым делать новые открытия.

Каркасы создаются путем связывания контигов вместе с использованием дополнительной информации об относительном положении и ориентации контигов в геноме. Контиги в каркасе разделены промежутками, которые обозначаются переменным количеством букв «N». Скаффолдинг часто используется для сборок с коротким чтением, чтобы понять фрагментированные сборки генома, содержащие короткие контиги. Однако есть три важных принципиальных недостатка строительных лесов:
- Леса пропускают важную информацию . Пробелы представляют собой недостающую геномную информацию, и во многих случаях эти пробелы могут совпадать с важными геномными локусами. Многие промоторы и первые экзоны имеют богатую GC последовательность, что часто приводит к отсутствующим или некачественным считываниям последовательности при коротком чтении или секвенировании по Сэнгеру. Таким образом, гены не полностью разрешены, и их регуляция не может быть понята. Еще одна причина пробелов в каркасных сборках — это большие повторяющиеся элементы, с которыми не удается справиться с помощью методов короткого чтения. Таким образом, дублированные гены, гены против псевдогенов, короткие тандемные повторы, тандемные повторы с переменным числом, микросателлиты и многие другие структурные геномные особенности часто остаются неразрешенными в каркасных сборках с коротким чтением. Как резюмировано в статье Nature Genetic Reviews , технологии секвенирования с длительным чтением — и особенно чтения HiFi — помогают преодолеть эти типы сложных областей, чтобы дать полную картину генетической изменчивости, в том числе в областях, ранее считавшихся трудноизвлекаемыми, таких как теломеры и центромеры. .
- Длина зазора каркаса часто не имеет отношения к истинному размеру зазора . В некоторых эталонных геномах промежутки произвольно устанавливаются на определенную фиксированную длину. Например, большинство зазоров в эталоне для зебрового вьюрка установлено на 100 Нс, а в эталоне на кукурузу версии 3 они установлены на 1000 Нс. Это означает, что в большинстве случаев истинная длина последовательности, представленная пробелом, отличается от установленного размера пробела, а иногда и на тысячи оснований. Неопределенность размеров промежутков в каркасах приводит к невозможности понять истинные пространственные отношения функциональных элементов в геномах и является недооценкой фактического объема недостающей информации. Совсем недавно эти старые эталонные сборки получили преимущества от секвенирования с длинным считыванием PacBio — см. Последние: зяблик-зебра и кукуруза.
- Последовательность каркасов, граничащих с промежутком, может быть низкого качества, а иногда и совершенно неправильной . Последовательности, окружающие пропуски, часто попадают в области, где технологии короткого считывания имеют недостатки из-за смещения GC или ограничений длины считывания. Это может привести к тому, что последовательность будет более низкого качества, а в некоторых случаях и полностью ошибочной. Например, из-за сложных повторяющихся структур в локусе IGH человека правый край разрыва в 50 000 Н в сборке коротких ридов содержит 1836 оснований фланкирующей последовательности, которая не имеет поддержки в hg19. ссылка на геном человека или сборка PacBio. В некотором смысле неправильная последовательность фланкирования в каркасах хуже, чем наличие «N» пробелов, поскольку эта ошибочная последовательность учитывается и включается в последующие анализы.
 Таким образом, дублированные гены, гены против псевдогенов, короткие тандемные повторы, тандемные повторы с переменным числом, микросателлиты и многие другие структурные геномные особенности часто остаются неразрешенными в каркасных сборках с коротким чтением. Как резюмировано в статье Nature Genetic Reviews , технологии секвенирования с длительным чтением — и особенно чтения HiFi — помогают преодолеть эти типы сложных областей, чтобы дать полную картину генетической изменчивости, в том числе в областях, ранее считавшихся трудноизвлекаемыми, таких как теломеры и центромеры. .
Таким образом, дублированные гены, гены против псевдогенов, короткие тандемные повторы, тандемные повторы с переменным числом, микросателлиты и многие другие структурные геномные особенности часто остаются неразрешенными в каркасных сборках с коротким чтением. Как резюмировано в статье Nature Genetic Reviews , технологии секвенирования с длительным чтением — и особенно чтения HiFi — помогают преодолеть эти типы сложных областей, чтобы дать полную картину генетической изменчивости, в том числе в областях, ранее считавшихся трудноизвлекаемыми, таких как теломеры и центромеры. . Неопределенность размеров промежутков в каркасах приводит к невозможности понять истинные пространственные отношения функциональных элементов в геномах и является недооценкой фактического объема недостающей информации. Совсем недавно эти старые эталонные сборки получили преимущества от секвенирования с длинным считыванием PacBio — см. Последние: зяблик-зебра и кукуруза.
Неопределенность размеров промежутков в каркасах приводит к невозможности понять истинные пространственные отношения функциональных элементов в геномах и является недооценкой фактического объема недостающей информации. Совсем недавно эти старые эталонные сборки получили преимущества от секвенирования с длинным считыванием PacBio — см. Последние: зяблик-зебра и кукуруза. ссылка на геном человека или сборка PacBio. В некотором смысле неправильная последовательность фланкирования в каркасах хуже, чем наличие «N» пробелов, поскольку эта ошибочная последовательность учитывается и включается в последующие анализы.
ссылка на геном человека или сборка PacBio. В некотором смысле неправильная последовательность фланкирования в каркасах хуже, чем наличие «N» пробелов, поскольку эта ошибочная последовательность учитывается и включается в последующие анализы.Информация, упущенная сборками каркасов с пробелами, усложняет и может препятствовать последующему анализу и пониманию, связанному с функциональной и сравнительной геномикой. с точки зрения непрерывности и полноты, и они часто требуют трудоемкой последующей работы для устранения пробелов, что увеличивает время и стоимость проектов.
Сборки каркасов PacBio для репрезентаций генома в масштабе хромосомы
Для еще более дальних связей генома, например, для соединения крупнейших сегментных дупликаций и повторяющихся областей, исследователи могут пойти еще дальше, добавив информацию об каркасах в сборку PacBio, часто что приводит к репрезентациям генома от теломер к теломере в масштабе хромосомы.